Introduction
Polymers in a melt can be viewed as long molecules which coil around each other and hinder each other's motions: they are "entangled". Entanglements occur because bonds between two adjacent atoms in a polymer chain can never be crossed by other such bonds. The goal of our study was to simulate and to understand the dynamical and rheological behaviour resulting from this entanglement effect. Because of its relative simplicity, we have chosen polyethylene (PE) as our primary system of interest.Microscopic simulations
First, we performed molecular dynamics (MD) simulations of a melt of relatively short C120H242 chains (see Fig. 1). Using the atomistic MD force fields, the uncrossability of bonds is automatically guaranteed because of the extremely high energy barier that has to be overcome if two bonds want to cross. Unfortunately, MD simulations are computationally very demanding. The rapid internal motions of the molecules make it necessary to calculate the interactions at femtosecond (10^-15 s) intervals. As a consequence, simulations of even the relatively small system of C120 chains took a few months to complete. From these simulations we learned that various dynamic quantities, as measured in the C120 system, can consistently be described by the Rouse model using a single set of fit parameters, provided the length scales involved are larger than the statistical segment length.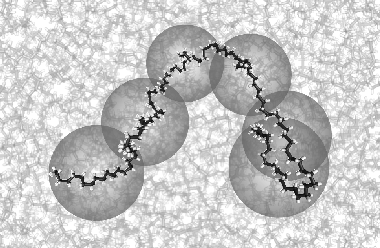
Fig. 1 In an atomistically detailed molecular dynamics simulation of a melt of C120H242 chains, we identify the centre-of-mass of each part of 20 carbon atoms as one "blob". In this picture one of the chains in the melt is highlighted.
Coarse graining
The longest characteristic time scale that occurs in a system of C120 chains is about 6 nanoseconds, which is well within reach of atomistic MD simulations. However, the longest time scale increases rapidly with chain length, and already equals several microseconds for a C1000 chain. Moreover, the system size must increase as well to avoid significant overlap of a chain with itself via the periodic boundary conditions. Hence, fully atomistic simulation of the dynamics and rheology of long polymer chains is quite impossible with current-day computer power. In order to increase the time and length scales accessible in simulations, it is necessary to describe the polymers on a more coarse-grained level. If the degree of coarse-graining is larger, a particular chain is represented by less coarse particles (which we call "blobs") and the integration time step can be increased. However, as long as the coarse-grained interactions are modelled as spherical interactions, it is important that the size of a blob does not exceed the entanglement length, since otherwise no realistic entanglement effects can arise. Taking these considerations into account, we decided to represent the center of mass of 20 consecutive CH2 groups by one blob.Thus, we derived the coarse-grained interactions between blobs from the underlying atomistic model (see Fig. 1). The resulting interactions are so soft that the uncrossability of bonds is no longer automatically met (Fig. 2). To prevent such unphysical bond crossings a new uncrossability constraint, the TWENTANGLEMENT algorithm, was introduced, details of which can be found in J. Chem. Phys. 115, 2846 (2001). The idea behind this constraint is to consider the bonds between consecutive blobs to be elastic bands. As soon as two of these elastic bands make contact, an "entanglement" is created at the crossing point which prevents the elastic bands from crossing (Fig. 3).
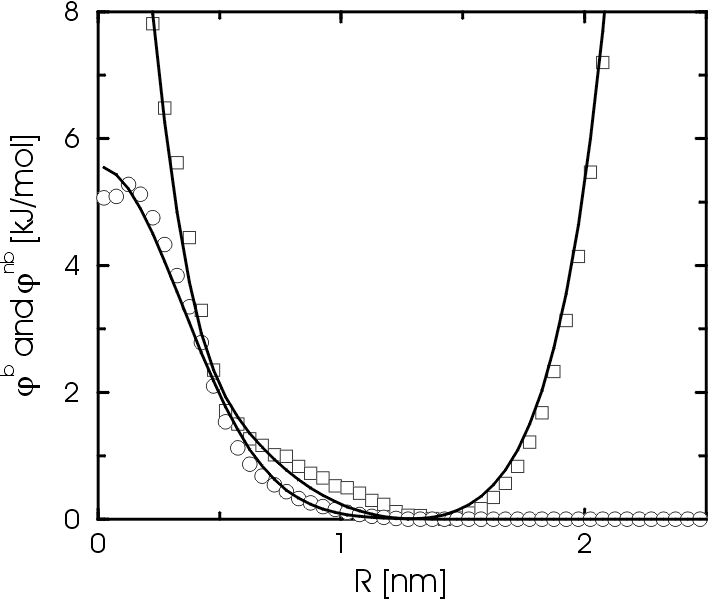
Fig. 2 Potential of mean force between bonded (squares) and non-bonded (circles) blobs. Notice that kT is about 3.7 kJ/mol at the temperature of 450 K.
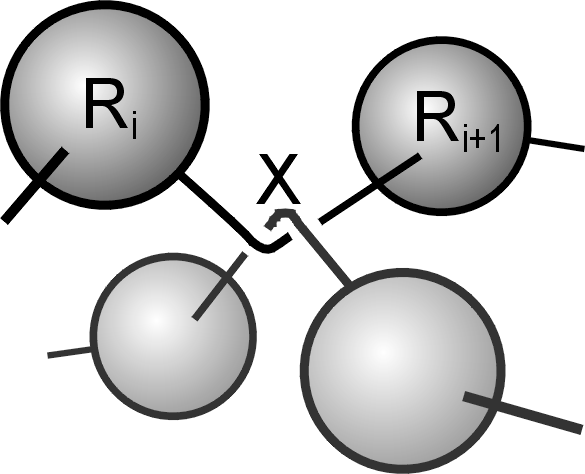
Fig. 3 Principle of TWENTANGLEMENT: at an earlier time, the bonds between the two depicted
pairs of blobs tried to cross each other. This caused the uncrossability constraint to insert an
"entanglement" at the crossing point. Since then the attractive part of the potential between
bonded blobs is a function of the path length from blob i, via the entanglement at X,
to blob i+1.
Results
The advantage of coarse-graining bottom-up, from the atomistic to the mesoscopic scale, is that all time and length scales are incorporated automatically and in their right proportions. Indeed, we found very good agreement between the dynamic results of the atomistic MD simulations of C120 and the coarse-grained simulations of B6. We observed a subdiffusive exponent in the mean square displacement of the chains, a stretching of the exponential decay of the Rouse modes, and a slowing down of the relaxation of the single chain coherent dynamic structure factor.Both the uncrossability of chains and their stiffness at smaller scales were found to be essential for these effects to occur. Interestingly, the shear relaxation modulus initially behaves Rouse-like, but after t=5 nanoseconds, the stress in the system relaxes more slowly than in a system of Rouse chains. This was attributed to a very slow interchain stress relaxation caused by the uncrossability of chains.
Next, we studied the dynamical and rheological behaviour of melts of chains ranging from C80 to C1000 (4 to 50 blobs). We found that the dynamics of chain lengths C400 - C1000 is in approximate agreement with reptation theory for large time scales, but that the approximation of a Rouse-like primitive path moving with great freedom in a tube is too strict. A better picture would be that of a primitive path that is interacting with the neighbouring chains on every length scale up to the entanglement length N_e. In fact we identified a new length scale, called the slowing down length N_s, which is smaller than the entanglement length N_e. The effective segmental friction increases rapidly around N_s leading, at constant density, to a transition in the scaling of the diffusion coefficient from D ~ N^-1 to D ~ N^-2 and a conspicious non-exponential relaxation behaviour. These effects were attributed to strong local kinetic constraints caused by both chain stiffness and interchain interactions. The onset of non-local (entanglement) effects occurs at a chain length of C120, as exemplified by deviations from Rouse behaviour of the shear relaxation modulus. Full (rheological) entanglement effects were observed only above C400, where the shear relaxation modulus displays a plateau (see Fig. 4) and the single chain coherent dynamic structure factor agrees with the reptation model. The results for the tube diameter and the plateau modulus, as well as diffusion coefficients and viscosities (Fig. 5) were found to be in good agreement with experiment.
Non-equilibrium coarse-grained simulations
Finally, as an application of our coarse-grained model, we studied the nonlinear flow properties of polyethylene melts by subjecting the model to a planar Couette flow. In steady state, typical effects such as shear thinning of the viscosity and a decrease of the extinction angle with shear rate were measured. Also transient effects, such as the characteristic overshoot in the shear stress and an undershoot in the transient extinction angle upon onset of shear flow, were measured and found to be in good agreement with experiments.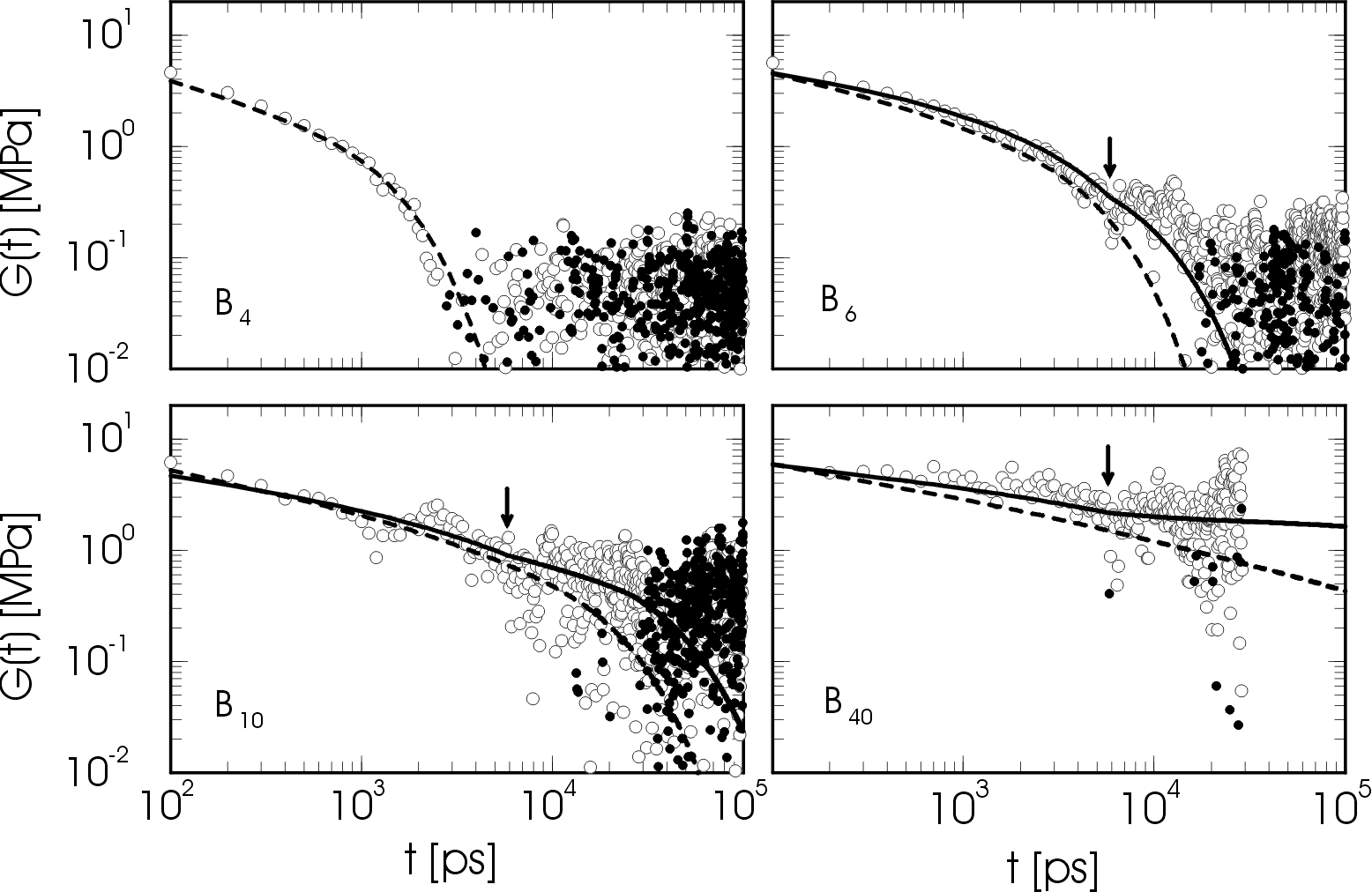
Fig. 4 Shear relaxation modulus of a melt of C80H162, C120H242, C200H402, and C800H1602, respectively. Absolute values of negative data are represented by filled circles. Dashed lines are Rouse model predictions with the measured spectrum of relaxation times. Solid lines are predictions from a mixed Rouse and reptation approach. The arrows indicate estimates of the entanglement time tau_e. Click on the picture for a larger view.
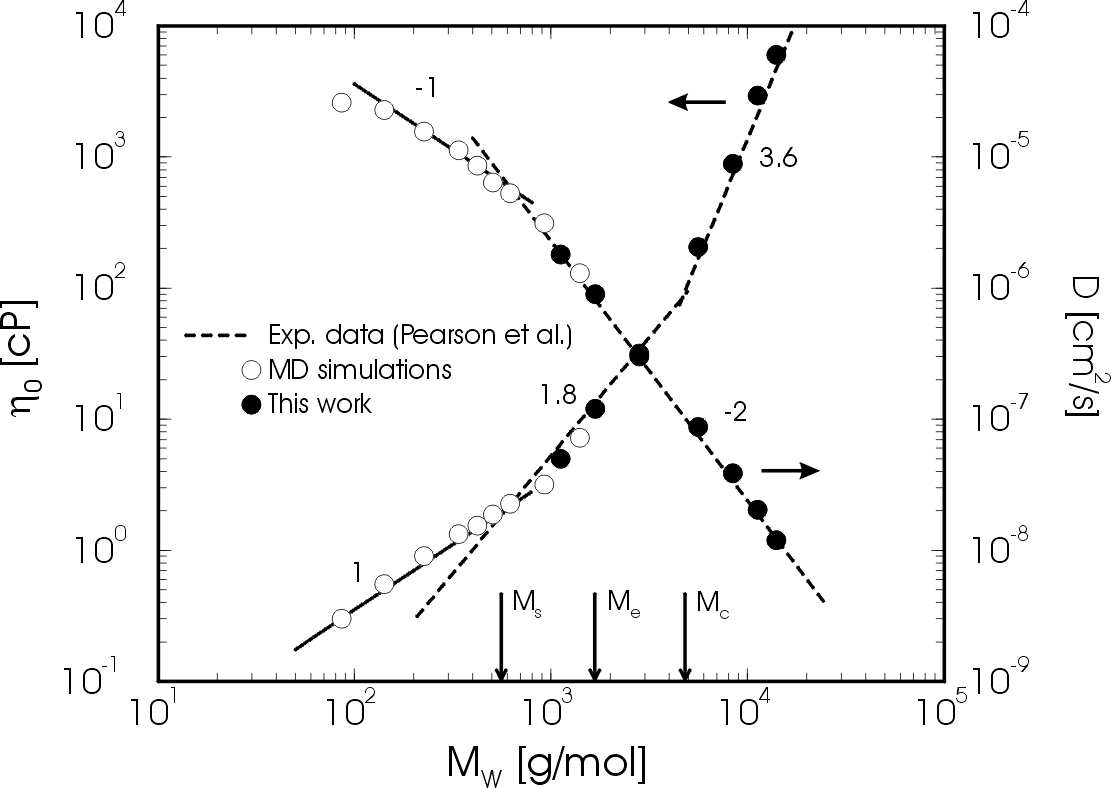
Fig. 5 Self-diffusion coefficient (descending curve; right scale) and viscosity
(ascending curve; left scale) versus molecular weight for polyethylene melts at 450 K.
Open circles are from atomistically detailed molecular dynamics simulations, closed circles
are from this work, and dashed lines are fits to experimental data.
Click on the picture for a larger view.